qPIPSA: Relating enzymatic kinetic parameters and interaction fields.
Razif R Gabdoulline, Matthias Stein and Rebecca C Wade
BMC Bioinformatics 2007, 8:373 (fulltext)
To follow this example you should firstly run the demonstration Part 1.
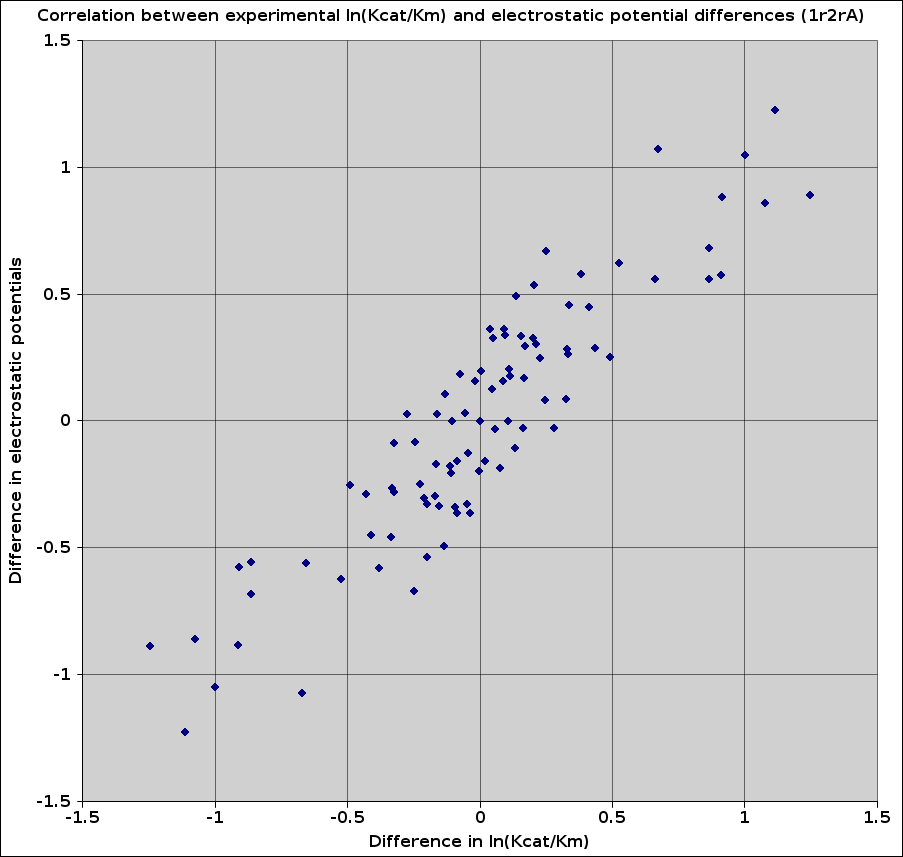
Plot the correlation between experimental ln(Kcat/Km) values and the electrostatic potential differences for TPI the x- and y- values are needed. Whereas the y-values are the differences in the electrostatic potentials and the x-values are the differences in ln(Kcat/Km).After completing the PIPSA run of Part 1 of the demonstration, you will find some *.log files in the job directory (see the email notification that you recieved after submitting the job).
Since in this example, 2 regions are specified:
you will find 3 *.log files:
which, respectively, contain the comparison for the two specified regions and for the whole protein.
The files contain a column with the differences in electrostatical potentials between all the proteins, which are used as the y-values in the plot (see the publication above for further details).
To extract the pertinent column out of the *.log files (e.g. sims.log) you can
In this demonstration the template used was the crystal structure 1R2RA. The correlation can alter depending on the template used to create the protein models. In Part 3 several templates have been used to examine the differences resulting from structural variations.